Thalassemia
Thalassemia
Thalassemia is described as a heterogeneous group of inherited anaemias characterized by a reduced or absent amount of haemoglobin. Thalassemia can be classified according to the deficient globin chain, alpha or beta thalassemia. The symptoms vary from relatively mild anaemia to life-threatening. Many studies have shown that reactive oxygen species are generated in increased amounts in thalassaemic red cells. Conditions such as rupture of erythrocytes, iron overload, and depletion of antioxidants in tissues (e. g. blood circulation) are considered in the promotion of oxidative stress. This implies the possible alteration of redox status in thalassaemic patients, which may adversely affect their health. Specific treatments for thalassemia are employed based on many factors such as age of the patients and severity of the disease.
Thalassemia is a blood disorder that affect the body’s ability to produce haemoglobin and red blood cells (RBC). People with Thalassemia have ‘lower than usual’ RBCs in their bodies and consequently low levels of the protein – haemoglobin. Lack of RBCs lead to infection, bone deformities, enlargement of the spleen and slower growth rates. Its treatment involves frequent blood transfusions, which can also lead to increased amounts of iron in the blood (although it can also happen because of the disease itself), which then, has to be dealt with medication. There are two main types of thalassemia, alpha thalassemia and beta thalassemia.
Alpha thalassemia: There are two genes (one from each parent) that produce alpha globin chains. Alpha thalassemia trait occurs if one or two of the four genes are affected.
Beta thalassemia: Two genes (one from each parent) are needed to produce sufficient beta globin protein chains. Beta thalassemia occurs if one or both genes are altered. The severity of beta thalassemia will depend on the number of affected genes. If both genes are affected it results in thalassemia major and severe anaemia.
Many people originating from the Mediterranean area, the Middle East, Africa or Asia carry thalassaemia.
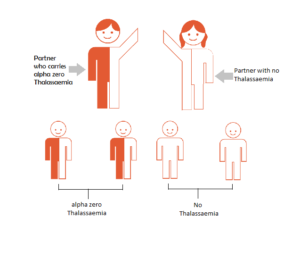
ALPHA THAL CARRIER AND NON CARRIER
Inheriting Thalassaemia – Alpha Thal Carrier & Non Carrier
If a couple are tested and only one partner proves to be a carrier, then there is no danger to their children. In each pregnancy there is a 1 in 2 chance that the child will carry alpha zero thalassaemia.
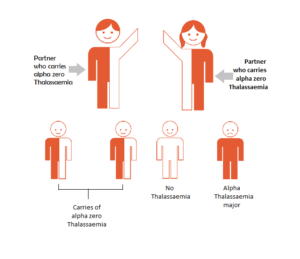
ALPHA THAL CARRIER & ALPHA THAL CARRIER
Inheriting Thalassaemia – Alpha Thal Carrier & Alpha Thal Carrier
If both partners are carriers, then there is a 1 in 4 chance of each pregnancy to produce a child with Alpha Zero thalassaemia major. This is a very serious anaemia that develops in the unborn baby and it can only happen when both parents carry alpha zero thalassaemia.
Genetic counselling is suggested to discuss the several options open for having a healthy family. If after full discussion the couple decide to undertake a pregnancy, the mother can be offered pre-natal testing for the disorder.
BETA THAL CARRIER & BETA THAL CARRIER
Inheriting Thalassaemia – Beta Thal Carrier & Beta Thal Carrie
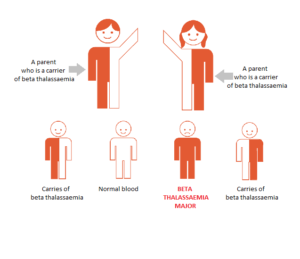
If a couple are tested and one partner proves to be a carrier, then there is no danger to their children (same as Fig 1). If both partners are carriers, there is a 25% chance of each pregnancy to produce a child with beta thalassaemia major.
Genetic counselling is suggested to discuss the several options open for having a healthy family. If after full discussion the couple decide to undertake a pregnancy, the mother can be offered pre-natal testing for the disorder.
Diagnosis is typically done by blood tests including a complete blood count, special haemoglobin tests and genetic tests. Diagnosis may occur before birth through prenatal testing.The other common blood disorders that can be detected by testing are Haemoglobinopathies and sickle cell anaemia.
Around 10 to 15,000 babies with Haemoglobinopathies are born in India every year. The burden of hemoglobinopathies in India is high with nearly 12,000 infants being born every year with a severe disorder. These numbers imply that every hour a child is born who will suffer with this genetic disorder. With respect to sickle cell anaemia, the average frequency of sickle cell gene, a group of blood disorders typically inherited from a person’s parents, is around 5% in India and the highest sickle cell gene in different states of India are among Orrisa, Assam, Madhya Pradesh, Uttar Pradesh, Tamil Nadu and Gujarat. The table no. 1 depicts the prevalence of sickle-cell disease across these states.
Table 1: Prevalence of sickle-cell anaemias across different states in India
| State | Population (%) |
| Orissa | 9.00% |
| Assam | 8.30% |
| Madhya Pradesh | 7.40% |
| Uttar Pradesh | 7.10% |
| Tamil Nadu | 7.10% |
| Gujarat | 6.40% |
It is estimated that there are 35 million carriers of Thalassemia i.e. 1 in every 25 Indians. The carrier rate for β – thalassemia varies from 1% to 17 % in India with an average of 3.2 %.5 Both alpha and beta thalassemia include the following two forms of Thalassemia condition:
Thalassemia minor
- Thalassemia minor is not a disease and there are no physical sign and symptoms of the disease, as it cannot be identified without a blood test.
- Thalassemia minor never gets transformed into thalassemia major.
- No medication or treatment is required for thalassemia minor. One should consult a doctor for further advice.
Thalassemia major
- Within 3 to 8 months of birth, signs of blood deficiency start to appear in a thalassemia major patient.
- The person suffering from this disease needs regular blood transfusion during his/her short life-span.
- Thalassemia major is a fatal disease. In spite of an expensive and long treatment, survival of the patient beyond 20 years is usually difficult.
There is a genetic, ethnic and regional diversity of haemoglobin variants as well as of mutations in India. The marriage of couples from the same ancestry may increase the chances of blood disorder traits in the coming generation. The prevalence of thalassemia is at a rate of 8.1%, 7.9% and 6.5% in the state is especially high in the Thakker community that is sub categorised as Bhanushali (Kutch), Bhatiya (Kutch) and Lohana (Saurashtra) communities, respectively against a state average.
Table 2 & 3 depict the prevalence of common haemoglobinopathies and beta (ß) – thalassemia trait in tribal and non-tribal districts of Gujarat.
Table 2: Prevalence of common haemoglobinopathies in tribal districts of Gujarat
| Districts | Number | ß-thalassemia trait (%) |
| Banaskantha | 9,489 | 423(4.45) |
| Baroda | 31,608 | 381(1.2) |
| Bharuch | 19,373 | 330(1.7) |
| Dahod | 15,975 | 410(2.57) |
| Narmada | 31,966 | 464(1.45) |
| Panchmahal | 23,077 | 422(1.83) |
| Sabarkantha | 37007 | 505(1.37) |
| Total | 1,68,495 | 2,935 (1.76) |
Source: National Journal of Community Medicine Vol 3 Issue 1 Jan-March 2012
Table 3: Prevalence of common haemoglobinopathies in non-tribal districts of Gujarat
| Districts | Number | ß-Thalassemia trait (%) | |
| Ahmedabad | 19,825 | 678 (3.40) | |
| Anand | 24,329 | 482 (1.98) | |
| Gandhinagar | 3,165 | 71 (2.24) | |
| Bhavnagar | 1,009 | 28 (2.77) | |
| Amreli | 468 | 14 (2.99) | |
| Districts | Number | ß-Thalassemia trait (%) | |
| Jamnagar | 864 | 30 (3.47) | |
| Kheda | 1,812 | 45 (2.48) | |
| Kutch | 2,476 | 102 (4.10) | |
| Navsari | 12,175 | 230 (1.89) | |
| Mehsana | 31,161 | 432 (1.36) | |
| Patan | 12,691 | 173 (1.36) | |
| Rajkot | 2,284 | 70 (3.10) | |
| Surat | 31,368 | 732 (2.33) | |
| Porbandar | 725 | 21(2.89) | |
| Surendranagar | 1,042 | 8 (0.77) | |
| Valsad | 3,625 | 145 (4.00) | |
| Total | 1,49,044 | 3,261 (2.18) | |
Source: National Journal of Community Medicine Vol 3 Issue 1 Jan-March 2012
Apart from blood transfusions and iron chelation therapy, there is no known cure for this blood disorder. Bone marrow transplantation is the only option for cure. However, even if the child is operated, the chances of survival are only 50%. Also, transplantation treatment is highly expensive and is out of reach for the most marginalized families. Hence, it is imperative that appropriate awareness, affordable screening and treatment facilities are made available for the prevention and cure of Thalassemia especially among the poor and marginalized.